
Artículos de revisión
Tejido adiposo como glándula endocrina. Implicaciones fisiopatológicas.
ADIPOSE TISSUE AS AN ENDOCRINE GLAND. PATHOPHYSIOLOGICAL IMPLICATIONS.
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2011-06-02 18:17:53
Aprobado: 2011-07-14 10:11:59
Correspondencia: Dayamí García Torres. dayami.garcia@gal.sld.cu
RESUMEN
El tejido adiposo se considera en la actualidad como un órgano con importante función endocrina; distinguiéndose dos tipos: el pardo y el blanco. El tejido adiposo es capaz de secretar diversas sustancias conocidas como adipocinas que se encuentran implicadas en la regulación del peso corporal, en el sistema inmune, en la función vascular y en la sensibilidad insulínica. En la obesidad, principalmente visceral, se encuentran aumentadas las adipocinas proinflamatorias. Estas se asocian a la inflamación clínica y subclínica, a la insulinorresistencia, al estrés oxidativo y a la lesión endotelial; desarrollan un papel clave en el síndrome metabólico e incrementan el riesgo cardiometabólico. El objetivo de la presente revisión bibliográfica ha sido exponer las características morfofuncionales del tejido adiposo y los mecanismos que lo vinculan con la génesis de diversas enfermedades.
Palabras clave: tejido adiposo; glándulas endocrinas; obesidad abdominal; índice de masa corporal; peso corporal
ABSTRACT
Key words: adipose tissue; endocrine glands; obesity, abdominal; body mass index; body weight
INTRODUCCIÓN
La consideración del tejido adiposo como reservorio de energía ha venido cambiando con los años y desde 1987 se lo consideró como el principal sitio de producción de esteroides sexuales.1 Posteriormente, en 1994, se identificó la leptina, una hormona producida principalmente en el adipocito.1 Desde entonces se ha descubierto una amplia variedad de moléculas con una gran actividad biológica producida y secretada por los adipocitos, denominadas adipocitoquinas, entre las que se encuentran el factor de necrosis tumoral alfa (TNF-α), la leptina, la resistina, la adiponectina, y el inhibidor-1 del activador del plasminógeno (PAI-1) entre otras.
El tejido adiposo puede considerarse formalmente como un tejido endocrino, ya que produce y secreta péptidos con diferentes efectos que ejercen su acción en tejidos distantes (efecto endocrino), en contraste con efectos locales (paracrino o autocrino). Las células endocrinas clásicamente son controladas por estímulos externos que generan un mecanismo de retroalimentación. Estudios experimentales y clínicos en modelos animales y en humanos, respectivamente, han demostrado que las hormonas y citocinas producidas por los adipocitos ejercen sus acciones en el sistema nervioso central, el músculo, el hígado, y el hueso entre otros muchos tejidos. El tejido adiposo también participa en los procesos de inflamación, regulación metabólica de energía, enfermedad vascular ateroesclerótica, síndrome metabólico, y cáncer.2,3
En la última década se ha reconocido el importante papel de los adipocitos en la homeostasis de energía corporal, la sensibilidad a la insulina, y el metabolismo de carbohidratos y lípidos.4
Para explicar la asociación entre adiposidad y enfermedad, se han postulado tres teorías: la portal/visceral, la lipodistrofia adquirida y la teoría del paradigma endocrino.
La primera otorga un papel central al aumento de grasa visceral y a su drenaje directo a la circulación porta, que lleva a una inhibición de la acción de la insulina, disminuyendo la oxidación de la glucosa y su utilización muscular, con lo que aumenta la producción hepática de glucosa y de lipoproteínas de muy baja densidad, además de un efecto lipotóxico sobre la célula beta, eventos todos que podrían explicar la relación entre obesidad, insulinoresistencia y diabetes de tipo 2.5
La teoría de la lipodistrofia adquirida (o síndrome de almacenamiento ectópico de grasa) se basa, primero, en la presencia de insulino resistencia severa y de diabetes, probablemente como consecuencia del almacenamiento de lípidos en el hígado, en el músculo y en las células beta pancreáticas, en pacientes con lipodistrofia; segundo, en la correlación entre insulino resistencia y el grado de infiltración lipídica en el tejido muscular esquelético, hígado y probablemente en las células beta en los pacientes obesos y, tercero, en el hecho de que el incremento en el tamaño de la célula grasa se asocia con insulina resistencia y diabetes, representando esta situación una incapacidad para expandirse y para acomodar un alto flujo de energía.6
Por último, la teoría del paradigma endrocrino se basa en el conocimiento del tejido adiposo como un órgano endocrino que produce péptidos bioactivos, que no sólo influencian al adipocito en una forma autocrina y paracrina, sino que afecta varias funciones metabólicas a distancia5 y en la presencia, en este tejido, de numerosos receptores que le permiten responder a diversas señales aferentes desde varios sistemas hormonales y el sistema nervioso central.
La importancia de estos hallazgos es aún mayor si tenemos en cuenta que el tejido adiposo constituye, en cantidad, el mayor órgano de la economía corporal, recibe una rica vascularización que asegura la circulación sistémica de los péptidos que produce y, sobre todo, tiene un potencial de crecimiento casi ilimitado a cualquier edad.7
A medida que la prevalencia del sobrepeso y de la obesidad se incrementan, también lo hace el interés por un mejor conocimiento del tejido adiposo, de tal forma que está demostrada la importancia de este tejido en enfermedades que se derivan de su exceso y de su deficiencia, las cuales se asocian con insulino resistencia, con hiperglucemia, con dislipemia, con hipertensión y con estados protrombóticos y proinflamatorios, componentes todos del síndrome metabólico, el cual se define como un grupo de factores de riesgo de origen metabólico que se acompañan de un riesgo aumentado de diabetes tipo 2 y de enfermedades cardiovasculares. Este síndrome está alcanzando proporciones epidémicas, calculándose que cerca de una cuarta parte de la población mundial lo padece actualmente.8
Este trabajo se centra en la descripción de las principales funciones endocrinas descritas para el tejido adiposo blanco y en su implicación fisiopatológica. Tiene como objetivos describir las características morfofuncionales del tejido adiposo, explicar las acciones de las sustancias secretadas por las células del tejido adiposo y explicar las implicaciones de las adipocinas en la génesis de diversas afecciones.
DESARROLLO
Morfofisiología del tejido adiposo
Histológicamente, el tejido adiposo, visto como un órgano, no está formado solamente por los adipocitos, ya que estos constituyen aproximadamente el 60-70 % de su estructura. El tamaño de los adipocitos puede variar considerablemente desde 20 a 200 micrómetros de diámetro, lo que significa que pueden en ciertas circunstancias aumentar hasta 1000 veces su volumen. El resto del tejido está constituido por células sanguíneas, células endoteliales, macrófagos, pericitos y precursores de los adipocitos en distintos grados de diferenciación, ya sean fibroblastos como precursores primarios, y preadipocitos, células intersticiales vacías de lípidos y prontas a transformarse en adipocitos.9
Los procesos celulares que llevan a la conversión de las células multipotentes en adipoblastos y posteriormente en preadipocitos y adipocitos, son todavía poco conocidos, aunque en la actualidad se reconocen mecanismos que permiten una aproximación con mayor certeza sobre lo que ocurre en la formación del tejido adiposo. Las principales funciones del tejido adiposo son: reserva energética, amortiguación-protección ósea y aislamiento térmico, aunque también ahora se acepta que posee funciones endocrinas y paracrinas.10
Desde el punto de vista histológico, el tejido adiposo blanco está altamente vascularizado (aunque menos que el tejido adiposo pardo), a tal punto que muchos adipocitos se encuentran en contacto directo con uno o más capilares. Estos permiten la entrada y salida activa de metabolitos, péptidos y factores no peptídicos, fundamentales en la regulación de la diferenciación y el crecimiento celular.11
Se ha logrado identificar dos etapas de crecimiento acelerado del tejido adiposo blanco, una después del nacimiento y otra que se produce durante el predesarrollo puberal, entre los 9 y 13 años de edad. La tasa de proliferación del tejido adiposo decrece en la adolescencia y se presenta un equilibrio relativo hasta la adultez. Normalmente la expansión del tejido adiposo se debe principalmente a hipertrofia de las células ya presentes (aumento del tamaño celular), sin un aumento en el número de células (hiperplasia). Sin embargo, una vez que se desencadenan los mecanismos que conducen a la obesidad, se produce un aumento no solo del tamaño de las células adiposas, también un aumento del número de células, las que pueden hasta quintuplicar el número original determinado en la postadolescencia.12
Generalmente el tejido adiposo se deposita en áreas con abundante tejido conectivo laxo, como por ejemplo las capas subcutáneas entre el músculo y la dermis. Sin embargo, también se localiza en forma típica, alrededor de las vísceras, riñones, corazón y otros órganos internos. Muchos estudios avalan el hecho de que el tejido adiposo no es un órgano homogéneo; más aún, se plantea que la ubicación topográfica del tejido adiposo hace que tenga perfiles metabólicos distintos dependiendo de la ubicación anatómica, lo cual lo hace susceptible de participar en el desarrollo de ciertas enfermedades.13
Tipos de tejido adiposo
Aún persiste una de las más antiguas clasificaciones del tejido adiposo y que tiene como base la coloración que este adquiere ante tinciones fundamentales utilizadas en anatomía patológica. Es así como se le clasifica en tejido adiposo blanco (TAB) y tejido adiposo pardo o marrón.
El tejido adiposo blanco, estructuralmente, es unilocular (de estructura intracelular uniforme). Cada adipocito contiene una gota central grande de triacilglicéridos (estructura unilocular) y el citoplasma queda reducido a un fino reborde de la célula. El núcleo, de estructura oval, desplazado hacia la zona periférica, contiene cromatina de grano fino y no presenta un nucléolo visible. El adipocito del tejido adiposo blanco posee pocas mitocondrias, un retículo endoplasmático rugoso y liso de baja densidad membranosa y un complejo de Golgi de pequeño tamaño. La gota de lípido está limitada por pequeños filamentos proteicos denominados perilipinas.14
El tejido adiposo blanco es muy vascularizado, cada célula está en contacto con al menos un capilar. Este tejido está subdividido en pequeños lobulillos por tabiques de tejido conectivo no muy definidos.11, 12
El tejido adiposo blanco se encuentra distribuido como grasa subcutánea (tejido adiposo subcutáneo) y como panículo adiposo en el mesenterio y en la zona retroperitoneal (tejido adiposo visceral).13
El tejido adiposo subcutáneo (TAS) es cuantitativamente el más importante, y constituye alrededor de un 80 % del total de la grasa corporal. Su metabolismo es más bien lento en comparación con el tejido adiposo visceral. Esto es, los procesos de lipogénesis y lipogenólisis, discutidos más adelante, son de poca relevancia. El tejido adiposo subcutáneo de la región abdominal tiene un comportamiento mixto en términos metabólicos ya que puede ser tan activo como el visceral. Su función más destacada es la de aislante térmico y de amortiguación mecánica. También se ha propuesto que ejercería un efecto de “amortiguación metabólica” ya que podría de alguna manera amortiguar el impacto de los excesos calóricos en el tejido adiposo visceral, aunque este aspecto no está aún del todo claro.15,16
El tejido adiposo visceral (TAV) también es conocido como tejido adiposo intraperitoneal. Se le subdivide en omental y mesentérico y se ubica en las regiones profundas de la cavidad abdominal rodeando las vísceras. Constituye el 5-10 % del tejido adiposo total en mujeres y hombres, respectivamente, siendo similar este porcentaje tanto en individuos de peso normal como en obesos.15 Las mujeres, en la edad adulta y postmenopausia tienden a aumentarlo más que los hombres.17
Posee, además, receptores para glucocorticoides, con lo cual las situaciones de estrés crónico tienen gran impacto sobre los depósitos de grasa en este tejido, pudiéndose producir estímulo de su acumulación, o por el contrario, de su movilización (aumento o baja de peso en condiciones de estrés físico y/o emocional). Desde el punto de vista vascular, está sujeto a drenaje portal, con lo cual los ácidos grasos que se liberan por lipólisis llegan directamente al hígado, constituyendo así un aporte directo de energía para el metabolismo general.18
Sin embargo, un exceso de drenaje de ácidos grasos desde el tejido adiposo visceral al hígado, también facilita el desarrollo de insulinorresistencia hepática, hiperinsulinemia, dislipemia e hiperglucemia, de modo que su asociación con una mayor prevalencia de estas y otras enfermedades ya es evidente.18
Existen numerosas diferencias entre los adipocitos del TAV y los del TAS. Clásicamente se ha considerado que los adipocitos del TAV poseen mayor actividad lipolítica que los del TAS.19 Sin embargo, observaciones recientes muestran que los adipocitos del TAS, en términos absolutos, tienen mayor actividad de lipoproteinlipasa y superior efecto lipolítico tras estimulación farmacológica. No obstante, la capacidad de respuesta lipolítica relativa respecto al nivel basal es superior en los adipocitos del TAV. Estos hallazgos son compatibles con una mayor sensibilidad del TAV a estímulos lipolíticos.16
La producción de citocinas proinflamatorias y generadoras de insulino resistencia (IR) como la interleucina (IL) 6 y el factor de necrosis tumoral alfa (TNF-α), así como del inhibidor del activador del plasminógeno tipo 1 (PAI-1), es superior en el TAV que en el TAS, mientras que éste genera más leptina y adiponectina. La producción de leptina aumenta conforme lo hace el tamaño del adipocito, lo que representa un mecanismo de autorregulación, ya que la hiperleptinemia reduce la ingesta, aumenta la actividad simpática y favorece la oxidación de ácidos grasos en los músculos.20
Los adipocitos del TAV tienen mayor capacidad de captación de glucosa que los del TAS, en probable relación con una mayor expresión de GLUT-4,21 lo que es un sustrato para alcanzar un almacenamiento de triglicéridos superior al del TAS.
El mayor grado de lipólisis del TAV favorece el flujo aumentado de ácidos grasos libres al hígado por vía portal, donde contribuyen a generar IR, esteatosis hepática y sus complicaciones metabólicas. La densidad de receptores de andrógenos y glucocorticoides en el TAV es superior a la del TAS,22 lo que es la base de su regulación endocrina. El tono estrogénico favorece la acumulación gluteofemoral de TAS, mientras que su desaparición en la menopausia promueve la de TAV.23 La menopausia incrementa el tamaño de los adipocitos del TAV y su actividad lipolítica como consecuencia de la IR, mientras que en el TAS no se producen cambios respecto a la situación premenopáusica. Así pues, la proporción entre las magnitudes de TAV y TAS aumenta tras la menopausia y la aproxima a la situación del varón. El efecto inhibidor de los estrógenos sobre la producción de IL-6 también se reduce tras la menopausia,24 a pesar de que el adipocito se convierta en una fuente estrogénica a través de la aromatización de andrógenos. Consecuentemente, los estrógenos, por este y otros mecanismos, pueden contribuir al efecto protector contra la enfermedad cardiovascular que presentan las mujeres premenopáusicas cuando se las compara con los varones de su misma edad, lo que explicaría que el tratamiento hormonal sustitutivo en la menopausia ejerza un efecto protector contra la acumulación de TAV.
Hay indicios de que la magnitud del TAV influye en la sensibilidad a la lipólisis de los adipocitos del TAS, que se ve elevada en pacientes con obesidad visceral,25 lo que contribuiría a aumentar el contingente circulante de ácidos grasos libres (AGL). Por otra parte, algunos factores de transcripción presentes en el TAV se encuentran también en adipocitos del TAS. Los preadipocitos del TAS poseen mayor capacidad que los del TAV para diferenciarse en adipocitos pequeños, que muestran alta insulinosensibilidad y acumulan triglicéridos y AGL, evitando su depósito en otros tejidos como músculo, hígado, páncreas o miocardio, por lo que ejercen un papel protector contra la esteatosis orgánica y la lipotoxicidad, que son esenciales en el desarrollo de las complicaciones de la obesidad. Posiblemente, cuando los adipocitos del TAS ven superada su capacidad de almacenamiento, se vuelven insulinorresistentes, aumenta su capacidad lipolítica, liberan AGL y permiten el aumento del TAV y sus complicaciones.
Los estudios comparativos de expresión génica en TAS y TAV llevados a cabo en pacientes con obesidad contribuyen a esclarecer la función de ambos compartimentos de tejido adiposo. Tanto el TAV como el TAS muestran sobreexpresión de genes relacionados con la inflamación en pacientes obesos.19
El tejido adiposo pardo se encuentra principalmente alrededor del cuello y en los grandes vasos sanguíneos del tórax en los neonatos, con la función de mantener la temperatura corporal; (1) luego, en la edad adulta, es reemplazado por el tejido adiposo blanco; no obstante, en los adultos se conserva tejido adiposo pardo, en pequeños cúmulos, dentro del tejido adiposo blanco.26
El tejido adiposo pardo permite realizar lo que se conoce como termogénesis adaptativa o facultativa, esto es la capacidad que tiene el organismo para responder al frío. A diferencia del tejido adiposo blanco, el tejido adiposo pardo es multilocular, o sea, está formado por múltiples gotitas citoplasmáticas de diferente tamaño que contienen triacilglicéridos. Sus células son más pequeñas que las del tejido adiposo blanco, pero contienen una cantidad considerablemente mayor de mitocondrias. Su color varía desde el dorado al marrón rojizo y sus células tienen forma poligonal, exhibiendo un citoplasma más abundante y granuloso. El núcleo es redondeado, está ubicado casi al centro y contiene gránulos de cromatina bastante gruesos y con un nucléolo visible. En el citoplasma se observan numerosas mitocondrias grandes y redondas que presentan crestas muy juntas. Los demás organelos se encuentran poco desarrollados.26
El tejido adiposo pardo aparece a la microscopía característicamente lobulado y puede parecer una glándula en cuanto a su aspecto macroscópico. Presenta muchos más capilares que el tejido adiposo blanco, y posee numerosas fibras nerviosas entre sus células. El color marrón está dado por los citocromos que forman parte de la cadena respiratoria de las numerosas mitocondrias que posee.18
Este tejido es estimulado por la noradrenalina secretada por el sistema nervioso autónomo, produciéndose la hidrólisis aumentada de triacilglicéridos de los adipocitos por activación de la lipasa hormona sensible (LHS), y luego la oxidación de ácidos grasos por beta oxidación mitocondrial que va acompañada de gran consumo de oxígeno, produciéndose calor durante este proceso. Una proteína identificada como termogenina actúa como un factor discordante entre la respiración mitocondrial y la fosforilación oxidativa, de modo que se forma menos ATP y se disipa mayor cantidad de calor.25
La termogenina pertenece a un grupo de proteínas discordantes colectivamente conocidas como UCP (siglas del inglés uncoupling protein), las que juegan una función muy importante en el control de la termogénesis, especialmente en los animales que hibernan. Las UCP son 5, la UCP-1 se encuentra solo en el tejido adiposo pardo, la UCP-2 se distribuye en varios tejidos (músculo, riñón, vísceras), la UCP-3 solo se encuentra en el músculo esquelético, y las UCP-4 y UCP-5 se ubican en el cerebro.12
Este tejido permanece en reposo de por vida, desde el punto de vista termogénico. Se ha propuesto que la reconversión de este tejido nuevamente en células pardas, o la transformación de células adiposas blancas en pardas, podrían constituir un interesante abordaje para el tratamiento de la obesidad mórbida. Sin embargo, aún falta mucho por conocer sobre la regulación y la biología molecular de estas células.18
Producción adipocitaria endocrina
Las adipoquinas, diversas en cuanto a su estructura proteica y a su función fisiológica, establecen una red de comunicaciones con tejidos y órganos como el músculo esquelético, la corteza adrenal, el cerebro y el sistema nervioso simpático.4 Incluyen citoquinas clásicas, factores de crecimiento y quimitotácticos, factores del complemento y proteínas comprometidas en la regulación de la presión arterial, la homeostasis vascular, el metabolismo lipídico, la homeostasis de glucosa, la angiogénesis y la osteogénesis. (Cuadro 1)

Leptina: La función del tejido adiposo, como emisor de señales eferentes por medio de péptidos, comenzó a acaparar atención tras la identificación y caracterización del gen de la leptina (LEP, 7q31.1) en el año 1994, si bien no fue el primer péptido de producción adipocitaria descrito y, posteriormente, de su receptor entre 1995 y 1996. Hasta la fecha es la adipokina mejor estudiada.28
Su nombre proviene del griego leptos (delgado). Es un polipéptido de 16 kDa que contiene 167 aminoácidos, con homología estructural a las citoquinas. Es secretada mayormente por los adipocitos, aunque también puede ser secretada por células inmunocompetentes y endoteliales, en proporción directa a la masa de tejido adiposo, al contenido de triglicéridos y al estado nutricional, siendo mayor en el tejido subcutáneo que en el tejido visceral y disminuyéndo su producción con el ayuno prolongado.29
Sus receptores, miembros de la superfamilia de receptores de citoquina clase I, se expresan en el sistema nervioso central y periférico, identificándose variantes de ellos, aunque al parecer es su forma primaria la que media la mayoría de sus efectos. (34) Las isoformas ObRa y ObRc son receptores cortos que se localizan predominantemente en la barrera hematoencefálica, donde parecen desempeñar un papel transportador, sobre todo el ObRa. La forma ObRb, estructuralmente más larga, contiene un dominio de señalización intracelular para la vía JAK/STAT (Janus activated kinase/signal transducer activator of transcription), abunda en el hipotálamo y se asume como el principal impulsor de las acciones de esta hormona. Por último, la isoforma ObRe es la fracción soluble, la cual transporta la leptina por el torrente circulatorio, regulando su depuración y su vida media.7
Este receptor se expresa en bajos niveles en numerosos tejidos, pero en el hipotálamo medio basal, particularmente, en el núcleo arcuato, en el núcleo ventromedial y en el dorsomedial, se encuentra en altos niveles. En el núcleo ventromedial del hipotálamo, la leptina estimula los receptores citoquin quinasa 2 (CK2), la síntesis de la hormona estimulante de los melanocitos y el transcriptor regulado por cocaína-anfetamina (CART), moléculas, que por vía paracrina, estimulan los receptores 3 y 4 de melanocortina del núcleo lateral provocando saciedad. A ese mismo nivel inhibe péptidos orexígenos, como el neuropéptido Y, y la proteína relacionada con el gen-agouti.30,31
La leptina inhibe la lipogénesis y estimula la lipólisis, reduciendo los niveles de lípidos intracelulares en el músculo esquelético, el hígado y las células beta pancreáticas, mejorando de esta manera la sensibilidad a la insulina. En el músculo, esta sensibilidad se logra mediante la inhibición de la malonil Co-A, lo que incrementa el transporte de ácidos grasos a la mitocondria para la beta oxidación. En el sistema límbico estimula la recaptación de dopamina, bloqueando así el placer de comer y a través del núcleo cerúleo, activa el sistema nervioso simpático que conlleva un incremento del gasto energético en condiciones de reposo.31
La principal acción de la leptina es la de actuar como señal de saciedad. Durante el ayuno la leptina desciende rápidamente y estimula también la liberación de glucocorticoides, la reducción de tiroxina, de las hormonas sexuales y de la hormona del crecimiento. Por su acción angiogénica puede afectar la estructura vascular, y por medio del receptor plaquetario de leptina contribuye a la trombosis arterial. In vitro estimula la producción de intermediarios reactivos del oxígeno como resultado de la activación de macrófagos.27
La expresión y secreción de leptina se incrementa por la insulina, por los glucocorticoides, por el factor de necrosis tumoral alfa (TNFα), por los estrógenos y por proteínas alfa de unión al activador CCAAT (enhancer-binding protein alfa). De igual forma se disminuye por la actividad adrenérgica -β3, por los andrógenos, por los ácidos grasos libres, por la hormona de crecimiento y por los agonistas del receptor nuclear PPARg (Peroxisome Proliferator- Activated Receptor Gamma).5
Es paradójico el hecho de que la leptina se encuentra sobreexpresada en el TAB en la mayoría de los obesos, lo que ha llevado al desarrollo del concepto de la resistencia a la leptina. Un mecanismo planteado de la resistencia a leptina puede ser la inducción del supresor de la señalización de citocinas 3 (SOCS-3), que puede inhibir la señalización intracelular de leptina. En la inflamación, la leptina actúa directamente sobre los macrófagos para aumentar su actividad fagocítica y la producción de citocinas proinflamatorias. También se ha involucrado a la leptina en la inflamación asociada con la aterosclerosis y el síndrome metabólico. La leptina actúa como una señal en la regulación de la sensibilidad a la insulina a nivel de todo el organismo. De manera más puntual, la resistencia a la leptina es en sí uno de los factores causales de las complicaciones cardiovasculares en la obesidad.32
Adiponectina: Esta hormona ha sido identificada por diferentes grupos y ha recibido diferentes nombres, entre ellos el de proteína de adipocito relacionada con complemento (Acrp30) por la homología con su factor C1q, así como adipoQ, APM1, GBP28. Se trata de una proteína 30kDa, codificada por el gen APM1 (3q27) compuesto por tres exones codificantes, expresada casi exclusivamente en el tejido adiposo blanco.33
Puede sufrir modificaciones postraduccionales de hidroxilación y glucosilación, produciendo trímeros, hexámeros, o isoformas de alto peso molecular. Posee un dominio amino terminal similar a colágeno, un dominio carboxiterminal globular que media multimerización, y una estructura terciaria que guarda alta similitud con el factor de necrosis tumoral alfa, a pesar de no tenerla en su estructura primaria.33 Está compuesta por una cola de colágeno y una cabeza globular, que forma dímeros y trímeros; estos complejos de alto peso molecular se encuentran en la circulación, aunque no se ha determinado cuál es la forma bioactiva.32
Estudios recientes, revelan que la administración de adiponectina recombinante, ya sea en su forma completa o en la forma aislada (cabeza globular), ejerce efectos hipoglucémicos y disminuye la resistencia a la insulina en modelos de ratones con obesidad o diabetes. También la adiponectina tiene propiedades antiaterogénicas al inhibir la adhesión de los monocitos a las células endoteliales y la transformación de macrófagos a células espumosas in vitro. El fenotipo del ratón con uno o más genes eliminados (ratón nocaut, del inglés knockout), para adiponectina, confirmó que existe un efecto protector mediado por adiponectina contra la aterosclerosis y la inducción de la resistencia a la insulina. El efecto sensibilizador de insulina por adiponectina es mediado, en parte, por un incremento en la oxidación de ácidos grasos a través de la activación de la adenosin monofosfato protein quinasa (AMPK) en el músculo esquelético; de manera similar a la acción de la leptina, la adiponectina activa a la AMPK en el hígado, y como resultado, disminuye la síntesis de glucosa por el tejido hepático.32
Para la adiponectina se han propuesto dos tipos de receptores: uno, con dos proteínas transmembranales similares y con homología a receptores de unión a proteína G conocidos como adipoR1 y adipoR2 y otro, la T-cadherina muscular, expresada primariamente en el músculo, y que funciona como receptor de alta afinidad para la adiponectina globular. AdipoR2 se expresa principalmente en el hígado y funciona como un receptor de afinidad intermedia para la adiponectina de alto peso molecular, habiéndose reportado una isoforma de alto peso molecular que promueve la actividad de la AMPK en los hepatocitos. De otra parte, sólo los trímeros activan la AMPK en el músculo, mientras que los hexámeros y la isoforma de alto peso molecular activan el factor nuclear kappa beta (NFkβ). Estas actividades divergentes se podrían explicar por diferencias en el patrón de expresión específica para cada tejido de forma tal que los efectos biológicos de la adiponectina dependen no sólo de sus concentraciones circulantes y de las propiedades de las diferentes isoformas, sino también de la expresión de tejido específico de los subtipos de su receptor.27
La adiponectina circula en concentraciones extraordinariamente altas, y representa cerca del 0,01 % de toda la proteína plasmática, al contrario de las otras adipoquinas. Sus niveles tienen correlación inversa con la masa corporal, con la insulino resistencia y con los estados inflamatorios. La interleucina 6 (IL-6) y el TNF-α son potentes inhibidores de su expresión y de su secreción (7) en las biopsias de TAB humano y en células en cultivo. Esto sugiere que la inducción de la resistencia a la insulina por TNF-α e IL-6 puede también ejercer una inhibición autocrina- paracrina de la liberación de adiponectina.32
La adiponectina inhibe la adhesión de monocitos a las células endoteliales, la transformación de macrófagos en células espumosas y la activación de células endoteliales. Ejerce también un efecto sinérgico con la leptina para mejorar la sensibilidad a la insulina.34 Dentro de sus efectos metabólicos se encuentran: la mejoría de la insulino sensibilidad a nivel hepático, el descenso del flujo de ácidos grasos libres no esterificados, el incremento de la oxidación de grasa y la reducción de la salida de glucosa hepática. En el músculo, la adiponectina estimula el uso de glucosa y la oxidación de los ácidos grasos.35
Se sugiere que los individuos con altas concentraciones de adiponectina son menos propensos a desarrollar diabetes tipo 2 que aquellos con concentraciones bajas, razón por la cual se le considera un importante marcador, tanto de resistencia a la insulina como de riesgo de enfermedad cardiovascular.36
Factor de necrosis tumoral alfa (TNF- α): Fue la primera proteína secretada por el tejido adiposo y por las células estromovasculares a la que se le descubrió un papel en la homeostasis de glucosa.37 El TNF-α es una proteína que se expresa como un péptido de 26 kDa en la membrana celular y sufre un corte que da lugar a su forma soluble de 17 kDa. Se expresa mayormente en el tejido subcutáneo comparado con el tejido visceral y ejerce sus acciones vía receptores de tipo I y II, expresados por el adipocito.33 El ARN mensajero (mARN) del TNF-α del tejido adiposo se correlaciona con el índice de masa corporal, con el porcentaje de grasa corporal y con la hiperinsulinemia.
A pesar de que en humanos los niveles del RNAm y de la proteína de TNF-α son bajos, en el TAB correlacionan positivamente con la adiposidad y disminuyen en sujetos obesos después de la pérdida de peso, mejorando la sensibilidad a la insulina. Un artículo reciente informa que en individuos obesos la mayor parte de la liberación de TNF-α en el tejido adiposo se presenta en las células no adiposas. A nivel molecular, TNF-α induce la lipólisis, activa las isoformas inflamatorias de las proteínas cinasas activadas por mitógenos (MAPK): la cinasa N-terminal de c-Jun y la de p38 MAPK y disminuye tanto la actividad del primer sustrato del receptor de la insulina (IRS-1) al inducir su fosforilación en residuos de serinas, como la expresión del transportador de glucosa (GLUT-4).32
En el tejido adiposo, el TNF-α reprime genes comprometidos en la captación de ácidos grasos no esterificados y de glucosa, suprime genes para factores de transcripción comprometidos en adipogénesis y lipogénesis, e interviene en la expresión de varios factores secretados por los adipocitos, incluyendo adiponectina e interleucina 6. Su efecto endocrino directo parece menos importante que los efectos indirectos, que resultan de la modulación auto y paracrina de los ácidos grasos no esterificados que disminuyen su captación por el tejido adiposo, así como la inhibición en la expresión de adiponectina y de IL-6.32
En la obesidad se han observado aumentos de la forma de TNF-α unida a la membrana (26 kDa), y esta forma puede actuar de manera autocrina alterando profundamente la biología del TAB. De manera local, el TNF-α aumenta la expresión de los genes del inhibidor del activador del plasminógeno (PAI-1) y C3 y disminuye la de adiponectina en el TAB. El TNF-α regula vías en el TAB, mediando, al menos en parte, las alteraciones en los niveles plasmáticos de otras adipocinas.32
Interleucina 6 (IL-6): Secretada por múltiples células, dentro de ellas células inmunes, fibroblastos, células endoteliales, músculo esquelético y tejido adiposo, se trata de una citoquina pleiotrópica con múltiples efectos, que van desde la inflamación y la defensa hasta el daño tisular.5 Circula de forma glicosilada, en tamaños que oscilan entre los 22 kDa a 27 kDa; de su receptor, homólogo al receptor de leptina, existe una forma transmembranal y otra soluble. Un complejo conformado por el receptor y por dos moléculas homodimerizadas comienza la señalización intracelular de IL-6, 1/3 de la cual se expresa en los adipocitos y en la matriz del tejido adiposo.38
El TAB humano produce grandes cantidades de IL-6 (10 a 30 % del total de la proteína circulante). Su expresión y secreción son de dos a tres veces mayores en el tejido visceral que en el tejido subcutáneo. Es posible que la mayor liberación de esta citocina se deba a las células del estroma vascular.32
Se correlaciona positivamente tanto con la obesidad como con la resistencia a la acción periférica de la insulina, y disminuye con la pérdida de peso. Constituye un predictor del futuro desarrollo de diabetes mellitus tipo 2 y enfermedad cardiovascular, lo que permite establecer una cuantificación fiable del exceso real de tejido adiposo.7
La IL-6 tiene un efecto directo sobre la sensibilidad a la insulina por varios mecanismos; después de ser secretada por el TAB en los depósitos viscerales, llega al hígado y puede estimular la secreción hepática de triglicéridos y la gluconeogénesis. Se sugiere también que IL-6 participa en la resistencia a la insulina alterando la señalización en los hepatocitos por la inducción de la proteína SOCS-3 (siglas en inglés de Supresor of Cytokine Signaling- 3), inhibiendo la autofosforilación del receptor de la insulina dependiente de insulina. Aunado a esto, en adipocitos de ratón disminuye también la activación del IRS-1 y la PI-3 cinasa, por lo que induce resistencia a la insulina. En modelos de roedores con diabetes, la IL-6 induce también resistencia a la insulina en el músculo y apoptosis en las células β. En conclusión, IL- 6 puede actuar a varios niveles, tanto de forma paracrina y autocrina en el tejido adiposo, como de manera endocrina, en los tejidos periféricos, alterando el peso corporal, la homeostasis energética y la sensibilidad a la insulina.32
Como en el sistema nervioso central los efectos de la IL-6 son diferentes y sus niveles se correlacionan negativamente con la masa grasa en los pacientes con sobrepeso, se sugiere una deficiencia central en la obesidad.38
Adipsina y proteína estimulante de acetilación: El TAB produce varias proteínas de la vía alterna del complemento, entre las que se encuentra la adipsina, que es una proteasa de serinas idéntica al factor D de esta vía. En este sentido, el TAB del humano libera gran cantidad de proteína estimulante de la acetilación (ASP), que es una proteína derivada del factor C3 que es conocido también como C3desArg.32 Ambas proteínas se correlacionan positivamente con adiposidad, con insulinorresistencia, con dislipemia y con enfermedades cardiovasculares.38
ASP promueve la captación de ácidos grasos por incremento en la actividad de la lipoproteina lipasa, promueve la síntesis de triglicéridos por aumento de la actividad de la diacilglicerol acil transferasa y desciende la lipólisis y la liberación de ácidos grasos libres en los adipocitos. También incrementa el transporte de glucosa en los adipocitos, incrementa la translocación de sus transportadores y mejora la secreción de insulina estimulada por glucosa. Un receptor para ASP unido a proteína G y conocido como C5L2, es expresado en los adipocitos.38
En el plasma de sujetos obesos se presentan aumentos sustanciales de ASP acompañados de una sobreexpresión moderada del RNAm de C3 en el TAB. No se sabe si el incremento en la concentración plasmática de ASP produce aumento en su actividad o resistencia. La resistencia a ASP podría promover la redirección del flujo de ácidos grasos libres del TAB al hígado.32
En el hepatocito, la ASP estimula el almacenamiento de triglicéridos a través de la estimulación del transporte de glucosa, el aumento de la reesterificación de ácidos grasos libres y la inhibición de la lipólisis. Sin embargo, su receptor y la vía de señalización no se han caracterizado.37
Visfatina: Fue identificada hace varios años como un factor estimulador de colonias de células pre B y los estudios preliminares sugieren que sus concentraciones están incrementadas en humanos con obesidad abdominal, con diabetes mellitus, o con ambas. Circula en concentraciones por debajo de las de insulina (<10 % en ayuno y <3 % en estado prandial), considerándose muy pequeño su papel en la capacidad de mantener niveles normales de glucemia. Como su expresión no es regulada por la alimentación ni por el ayuno, se considera un factor poco probable en la vía de señalización del receptor de insulina. Existen aspectos pendientes por aclarar, como el hecho de que los niveles séricos de visfatina están variablemente correlacionados con la diabetes tipo 2 y con otros estados de insulino resistencia. El incremento de su síntesis, asociado con obesidad, podría ser una repuesta compensatoria para tratar de mantener la glucemia.39
Se ha demostrado que la visfatina tiene actividad enzimática de nicotinamida fosforibosiltransferasa, con residencia en el núcleo y el citosol, sin que exista claridad acerca de si hay una secreción regulada de visfatina, o si sus niveles séricos reflejan un escape desde las células muertas o dañadas.39 Son varios los receptores que regulan su síntesis, la cual es estimulada por glucocorticoides e inhibida por TNF-α, por IL-6, por hormona de crecimiento y por agonistas de receptor β-adrenérgico.40,41 Terapéuticamente, los agonistas PPARg muestran efectos diferentes sobre su modulación y se ha demostrado recientemente que el fenofibrato estimula su producción.
Omentina: Se trata de otro péptido secretado principalmente por la grasa visceral y contrario a la visfatina; al parecer, se produce más en células estromales vasculares dentro de la grasa, que en los propios adipocitos. Aunque el mecanismo de acción de la omentina, incluyendo los tejidos blancos, su receptor y sus vías de transducción, están por esclarecerse, es interesante resaltar que la omentina se produce en cantidades considerables en el tejido adiposo de humanos y de macacos, pero no en el de ratones.42
Apelina: Es un ligando del receptor huérfano AJP que actúa básicamente de forma paracrina y secretado como un prepropéptido de 77 aminoácidos que da origen a péptidos de 36, de 17 y de 13 aminoácidos, siendo los dos últimos las formas más activas.39 La única forma de metabolismo que se le conoce es a través de la enzima convertidora de angiotensina 2, limitada principalmente al endotelio de arterias, arteriolas y vénulas del corazón, riñón, epitelio tubular renal y testículos, la cual cliva la fenilalanina terminal de las adeninas 13 y 16 y las convierte en péptidos inactivos. El receptor de apelina se expresa ampliamente en el cerebro y en casi todos los tejidos periféricos, especialmente en células endoteliales a nivel de vasos intramiocárdicos, renales, pulmonares, adrenales y endocárdicos.39 La apelina produce vasodilatación dependiente de endotelio, mediada por óxido nítrico y vasoconstricción independiente de endotelio, por su acción sobre las células musculares lisas vasculares, siendo la primera acción la que prima.39 Se ha descrito también su papel en angiogénesis, el incremento en la contractilidad miocárdica, pero sin aumento del gasto cardiaco (debido a la venodilatación que disminuye la precarga) y sin producción de hipertrofia a largo plazo. Produce también inhibición en la secreción de hormona antidiurética, lo que le daría una función en el manejo de fallo cardiaco. Sobre las hormonas pituitarias, luego de su administración, se ha visto: aumento de la hormona adrenocorticotropa y cortisol, supresión de prolactina, hormona folículoestimulante, hormona luteinizante, hormona estimulate de la tiroides y un efecto neutro sobre la hormona de crecimiento. Como adipoquina, se produce de manera proporcional con la cantidad de grasa y tiene propiedades anorexígenas, acompañadas de aumento de la temperatura corporal y la actividad locomotora. Además, inhibe la secreción de glucosa dependiente de insulina.45
Resistina: También conocida como ADSF (siglas del inglés de Adipose Tissue Specific Secretory Factor). Esta adipocina pertenece a una familia de proteínas de secreción ricas en cisteína en el dominio C-terminal llamadas FIZZ (siglas del inglés de Found Inflammatory Zone), ahora conocida como Retn. El RNAm de la resistina codifica para un polipéptido de 114 aminoácidos, con 20 de ellos como péptido señal y que es secretado en forma de dímero de 108 aminoácidos.32
Este péptido es producido, fundamentalmente, por las células mononucleares de la matriz estromovascular. Hay varias formas multiméricas de resistina que circulan en el plasma y su acción celular parece depender de las formas con menor peso molecular, que son dímeros unidos por un puente disulfuro.
La resistina se expresa en células inmunocompetentes como el monocito-macrófago. La proteína humana presenta 59 % de homología con el ratón, su expresión en el TAB humano es menor a la de los ratones y no está del todo claro cuál es su papel en el desarrollo de la resistencia a la insulina en humanos. Estudios recientes han informado que tanto la expresión de la resistina en el TAB como sus concentraciones séricas, se encuentran aumentadas en individuos obesos y con DM tipo 2.32
Existe considerable controversia alrededor del papel de esta proteína en los humanos. Numerosos estudios en humanos han fallado en demostrar una correlación clara y consistente entre la expresión de resistina en el tejido adiposo, o entre los niveles circulantes de resistina y la adiposidad o la insulinorresistencia.1 Pero lo que sí parece probado es que constituye un vínculo con el entorno inflamatorio, debido a su predominante producción por monocitos y por su correlación con los niveles de IL-6.46
Proteína 4 ligante de retinol (RBP-4): Es un miembro de la superfamilia lipocalina y está regulada por cambios en los niveles del transportador 4 de glucosa (GLUT-4) en los adipocitos. Su expresión aumentada desmejora la acción de la insulina en músculo y en hígado, de forma tal que sus niveles elevados se asocian con insulino resistencia en humanos obesos y en aquellos con diabetes tipo 2, así como en pacientes delgados no diabéticos, pero con historia familiar de diabetes.47 Su mecanismo de acción no ha sido dilucidado y no está claro si el proceso compromete un ligando retinoide, o algún otro mecanismo.48
Ácidos grasos no esterificados (NEFA): Conocidos desde tiempo atrás como productos de secreción del adipocito, se liberan primariamente durante el ayuno como fuente de nutrición para el resto del cuerpo, con acciones adicionales en la homeostasis de la glucosa.27
Un nivel elevado de NEFA inhibe la habilidad de la insulina para promover, tanto la toma periférica de glucosa en músculo y en grasa, como para reducir la producción hepática de glucosa. Los niveles elevados de NEFA de manera transitoria, como ocurre durante una ingesta de comida, tienden a aumentar la secreción de insulina, mientras que las elevaciones crónicas, como en los casos de insulinorresistencia, tienden a reducir la secreción de esta hormona. El efecto neto de estas acciones es promover la utilización de lípidos como una fuente de energía para la mayoría de tejidos, mientras que para las neuronas y para los glóbulos rojos, que dependen de la glucosa como fuente de energía, se reservan los carbohidratos. Se han propuesto varios mecanismos para explicar estos efectos, dentro de ellos la activación de la proteína quinasa C (PKC), el estrés oxidativo, la formación de ceramida, y la activación del sistema inmune innato vías estas que no se excluyen entre sí y que probablemente funcionen de manera interdependiente.27
Debido a que la lipólisis en los adipocitos es reprimida por la insulina, la insulino resistencia de cualquier origen puede llevar a la elevación de NEFA, la cual, a su vez, induce insulino resistencia adicional, como parte de un círculo vicioso. Este efecto está mediado por varios mecanismos, incluyendo la apoptosis inducida por lipotoxicidad en las células de islotes. En las células beta que escapan de la apoptosis, los NEFA descienden la “percepción” de glucosa mediante una inducción de la expresión de la proteína desacopladora 2, la cual desciende el potencial de membrana mitocondrial, la síntesis de ATP y la secreción de insulina.27
Inhibidor del activador del plasminógeno (PAI-1): Es una proteasa de serinas que inhibe al activador de plasminógeno, cuya función es dar origen a la plasmina para activar la cascada fibrinolítica. Se expresa en múltiples tipos celulares dentro del TAB y principalmente las células del estroma vascular y los preadipocitos de la grasa visceral son la principal fuente de PAI-1 plasmático.48
Su expresión y secreción son mayores en el tejido visceral que en el tejido subcutáneo y sus niveles elevados, observados en la obesidad y en la insulinorresistencia, se correlacionan positivamente con el síndrome metabólico. La pérdida de peso, la restricción calórica, el ejercicio y la mejoría de la sensibilidad a la insulina, con un tratamiento a base de metformina o de tiazolidindiona, reduce significativamente sus niveles circulantes.48
Recientemente se ha observado que en la obesidad el deterioro del sistema fibrinolítico participa en las complicaciones cardiovasculares, defecto asociado con la presencia de altas concentraciones del PAI-1, el cual es principal inhibidor de la fibrinólisis.48 En el TAB y otros tejidos, el PAI-1 afecta la migración celular y la angiogénesis mediante la competencia con vitronectina por el sitio de unión para integrina en la matriz extracelular, efecto observado in vitro para la migración de los preadipocitos. Es a través de esta función que el PAI-1 puede afectar el crecimiento del TAB. También se ha observado que la sobreexpresión del RNAm del PAI-1 en el TAB disminuye la hipertrofia en ratones sometidos a una dieta rica en grasa. En ratones genéticamente obesos la inhibición del gene del PAI- 1 reduce la adiposidad pero no tiene ningún efecto significativo sobre la masa del TAB en el modelo de inducción de obesidad por dieta. En conjunto estas observaciones sugieren que en la obesidad el aumento en la secreción del PAI-1 por el TAB, aunque inhibe la fibrinólisis, ejerce un efecto protector contra el crecimiento excesivo del TAB. Por último, en la resistencia a la insulina, la secreción del PAI-1 por el TAB se encuentra aumentada, ligando a la enfermedad vascular con la resistencia a la insulina, la obesidad y la DT2.32
Sistema renina-angiotensina-aldosterona: Varias de las proteínas de este sistema, entre ellas la renina, el angiotensinógeno (AGE), la angiotensina I, la angiotensina II, los receptores de angiotensina tipo 1 (AT1) y tipo 2 (AT2), la enzima convertidora de angiotensina y otras proteasas capaces de producir angiotensina II (quimasa, catepsina D y G), son producidas en el tejido adiposo. La expresión de angiotensinógeno, de enzima convertidora de angiotensina y de receptores AT1, es mayor en el compartimiento visceral que en el subcutáneo, 9 y la adiposidad se correlaciona con la angiotensina plasmática, con actividad de renina plasmática, con actividad plasmática de enzima convertidora de angiotensina.
El angiotensinógeno es producido principalmente en el hígado y su mayor fuente extrahepática es el tejido adiposo. Un aumento en su producción contribuye a un incremento de masa adiposa, porque según se cree, la angiotensina II actúa localmente como factor trófico para la formación de nuevo tejido adiposo.
De otra parte, la angiotensina II, que se une a sus receptores en adipocitos, en células estromovasculares y en terminales nerviosas, afectando la fisiología del tejido adiposo por alteración del flujo sanguíneo y por la actividad del sistema nervioso simpático, es capaz de promover directamente el crecimiento y la diferenciación del adipocito al promover la lipogénesis, e indirectamente, al estimular la síntesis de prostaglandinas; además, la angiotensina II regula la expresión de factores endocrinos tisulares como la prostaciclina, el óxido nítrico, el PAI-1 y la leptina;50 la proveniente de adipocitos maduros, inhibe el reclutamiento posterior de preadipocitos.
11 β-dehidroxiesteroide dehidrogenasa: Esta enzima, de la que se han descubierto dos isoformas, cada una con propiedades y con roles biológicos únicos y localizadas en el retículo endoplásmatico, cataliza la interconversión del cortisol activo a cortisona inerte. La isoforma 1, que regenera el cortisol metabólicamente activo de la cortisona, está incrementada en los sujetos obesos y la isoforma 2, que inactiva el cortisol, protege tejidos claves. Varias observaciones asocian la actividad de la enzima tipo 1 con obesidad, con insulinorresistencia y con otros hallazgos de síndrome metabólico, sin embargo, no se han encontrado diferencias en su actividad entre pacientes obesos con diabetes tipo 2 y sus controles obesos, lo que sugiere que su desregulación se asocia más con obesidad que con diabetes tipo 2.51
Implicaciones fisiopatológicas: Inicialmente, la idea de que el tejido adiposo pudiese tener un considerable efecto en el control glucémico global no fue fácil de aceptar, y los estudios iniciales determinaron que el tejido adiposo sólo representaba una fracción de la distribución después de una comida (cerca del 10 % al 15 %), siendo captada por el músculo la mayor parte. Sin embargo, se fue haciendo evidente que las alteraciones en la adiposidad tenían profundas implicaciones en la homeostasis de la glucosa: demasiada grasa (obesidad) y muy poca (lipodistrofia) se asociaban con insulinorresistencia y con hiperglucemia. Adicionalmente, los ligandos de PPARg tenían una actividad antidiabética excelente, a pesar de que la mayor parte de ellos se encontraban en el tejido adiposo y no en el músculo. Posteriormente, ha quedado evidenciado que el profundo efecto de los adipocitos en el balance de glucosa está mediado por varios mecanismos, dentro de ellos el papel vital de las adipoquinas, ya sea ejerciendo efectos antihiperglucémicos como lo hacen la leptina, la adiponectina, la visfatina y la omentina, o bien, con efectos pro hiperglucémicos como lo hacen la resistina, el TNF- α, la IL-6 y el RBP4.27
Inflamación, insulinorresistencia, estrés oxidativo, disfunción endotelial y riesgo cardiovascular: El tejido adiposo de los pacientes obesos se caracteriza por hipertrofia e hiperplasia de los adipocitos y por cambios en sus funciones metabólicas; está demostrado que el adipocito es el mayor productor de adipoquinas inflamatorias en estas condiciones.52 Un mecanismo mediante el cual se induce esta producción es el estrés del retículo endoplásmico (entendido como un aumento de sus demandas de funcionamiento) inducido por la obesidad, lo que ocasiona cambios en la arquitectura, aumento en la síntesis de proteínas y de lípidos y perturbaciones en los flujos de energía y de nutrientes intracelulares en el tejido adiposo, que llevan a la activación de diferentes vías de señalización, entre ellas, las quinasas de la región aminoterminal del factor de transcripción c-jun (JNK), del factor complementario inhibidor I- κβ (IKK) y de PKC, induciendo (por regulación post transcripcional) la producción adicional de mediadores inflamatorios.
Se ha observado que los adipocitos y varias células del sistema inmune, tales como las células T y los macrófagos, poseen características similares en cuanto a la producción de citocinas proinflamatorias y a las vías de señalización. Por ejemplo, las células precursoras de los adipocitos (preadipocitos) poseen potente actividad fagocitaria.53 Aunado a esto, muchos genes críticos para los adipocitos, incluyendo aquellos que codifican para factores de trascripción, citocinas, moléculas inflamatorias, transportadores de ácidos grasos y receptores basureros, (scavenger) también son expresados en los macrófagos y tienen un papel importante en la biología del macrófago. Los preadipocitos pueden sufrir una transdiferenciación y adquirir la capacidad fagocítica, al ser introducidos en el peritoneo expresando F4/80, Mac-1 y CD45, pero este fenómeno es dependiente de la presencia de macrófagos.
Otro mecanismo que también puede ser relevante en la iniciación de inflamación, es el estrés oxidativo que se produce como consecuencia del incremento en el aporte de glucosa al tejido adiposo: al aumentar las células endoteliales, la captación de glucosa genera un aumento en la producción de radicales superóxido a nivel mitocondrial que ocasiona daño oxidativo, incremento en la producción de citoquinas inflamatorias y activación de las cascadas de señalización inflamatoria dentro de la célula endotelial.
La obesidad abdominal se asocia a disfunción endotelial, manifiesta por alteración de marcadores bioquímicos como moléculas de adhesión, trombomodulina y endotelina- 1. Como los adipocitos de pacientes obesos, también presentan una menor densidad de receptores de insulina y una mayor de receptores beta-3 adrenérgicos; se incrementa entonces la tasa de lipólisis con liberación de ácidos grasos libres, situación que tiene varias consecuencias metabólicas, dentro de ellas el aumento en la producción de radicales libre de oxígeno, la inducción de insulino resistencia, el sinergismo en la acción de la IL-6 y el TNF-α, y la inducción de apoptosis en la célula beta pancreática, efectos todos categorizados como de lipotoxicidad.31
Tanto el TNF-α como las IL, especialmente la IL-6 producida por los macrófagos, fibroblastos, células endoteliales y adipocitos y estimulada por el sistema nervioso simpático, son de los principales mediadores del aumento de proteínas inflamatorias. Su concentración elevada se asocia con resistencia insulínica y disfunción endotelial y predice diabetes mellitus tipo 2 e infarto de miocardio.
Aunque la administración de IL-6 inhibe la respuesta de insulina a la estimulación por glucosa, el efecto diabetógeno proviene sobre todo de su efecto inhibidor de la señalización intracelular del receptor de insulina en hepatocitos. También promueve el aumento de AGL y la reducción de adiponectina, circunstancias ambas que favorecen la IR.
Tanto la disminución de adiponectina, como el aumento de AGL y la interferencia con la señalización intracelular de insulina, a través de la reducción de la actividad de la tirosincinasa, son mecanismos potenciales del efecto en la IR del TNF-α. El TNF-α da lugar a cambios proinflamatorios en células endoteliales y de músculo liso vascular y estimula la producción de moléculas de acción vasoconstrictora como endotelina-1 y angiotensinógeno, lo que contribuye al desarrollo de hipertensión arterial. El TNF-α periarteriolar tiene un potencial papel patológico, que, en un efecto paracrino, determina un descenso en la producción de óxido nítrico que ocasiona una vasoconstricción arteriolar mantenida. De hecho, la concentración de TNF-α se relaciona con la presión arterial y la IR en humanos, por lo que constituye un elemento humoral clave en la fisiopatología del síndrome metabólico. Además, estimula la producción de IL-6 y PCR, con lo que se cierra un circuito reverberante generador de inflamación e IR.
Uno de los mecanismos por los que las adipocinas promueven la inflamación, es la estimulación de la producción hepática de PCR. Además de ser un marcador de inflamación, la PCR contribuye directamente a generar daño vascular, activación endotelial y trombosis. Se ha demostrado su capacidad de inhibir la sintetasa del óxido nítrico y de aumentar las moléculas de adhesión y el NF-kβ, el estrés oxidativo y la apoptosis endotelial, por lo que es un factor proarteriosclerótico. Las relaciones funcionales del estrés oxidativo con la activación del factor de transcripción NF-kβ, configuran uno de los mecanismos más importantes en la fisiopatología del riesgo cardiovascular propio del síndrome metabólico que se produce en el tejido adiposo.
La adiponectina, en contraste con el resto de las adipocinas, se encuentra reducida en la obesidad. Tiene efecto insulinosensibilizante, antiinflamatorio y antiaterogénico. La hipoadiponectinemia se asocia con la existencia de síndrome metabólico con mayor fuerza que cualquier marcador inflamatorio.54
Diversas investigaciones han demostrado que la adiponectina ejerce efectos directos sobre el endotelio vascular como disminución de la respuesta inflamatoria por lesión mecánica del endotelio, o bien de protección en el caso de deficiencia de apolipoproteína E.55 En relación a otros lípidos, diferentes genes vinculados con los niveles circulantes de adiponectina han mostrado efectos genéticos pleiotrópicos sobre las concentraciones de triglicéridos y de colesterol HDL.
Las concentraciones plasmáticas de adiponectina son menores en individuos con enfermedad coronaria comparados con controles pareados para edad y grado de obesidad. Los niveles de adiponectina también se encuentran disminuidos en personas con hipertensión arterial, independientemente de la presencia de resistencia a la insulina. Los individuos con niveles bajos de adiponectina presentan disminuida la vasodilatación dependiente de endotelio, lo que podría ser uno de los mecanismos implicados en la hipertensión arterial asociada a la obesidad central. Diferentes estudios han puesto en evidencia que la adiponectina produce efectos antiescleróticos directos. La adiponectina inhibe de manera importante la producción de moléculas de adhesión, como la molécula 1 de adhesión intracelular, la molécula 1 de adhesión celular vascular, y la selectina E, así como la proliferación de células musculares lisas inhibiendo la AMPK. La adiponectina también inhibe la activación del factor nuclear kβ inducido por el factor de necrosis tumoral-α, a través de la inhibición de la fosforilación del kβ. La supresión del factor nuclear kβ, mediada por la adiponectina, pudiera ser un importante mecanismo molecular de la inhibición de la adhesión de los monolitos a las células endoteliales.
La adiponectina también inhibe la producción de los receptores clase A-1 de los macrófagos, de lo que resulta una importante disminución de lipoproteínas oxidadas de baja densidad, lo que inhibe la formación de células espumosas.
Estudios con técnicas de inmunohistoquímica muestran que la adiponectina no se incorpora a la pared vascular normal intacta, mientras que se ha observado una marcada adhesión a las paredes vasculares previamente lesionadas de forma mecánica mediante catéteres con balón, ya que la adiponectina tiene la capacidad para unirse a los diferentes subtipos de colágena subendotelial, las lesiones endoteliales podrían inducir que la adiponectina penetrara al espacio subendotelial. Queda por establecerse con detalle la forma en que la adiponectina interactúa con las células del endotelio vascular, y el papel que pueden tener en la génesis las alteraciones vasculares.56
El mecanismo implicado en el efecto reductor de adiponectina, que tiene lugar en condiciones de obesidad, no se conoce bien, aunque se ha propuesto que la hiperproducción de TNF-α por el tejido adiposo visceral tiene efecto inhibidor de la síntesis de adiponectina por los adipocitos del tejido subcutáneo, lo que justifica los hallazgos en plasma.
Tanto la IL-6 como los glucocorticoides, inhiben la secreción de adiponectina. La hipoadiponectinemia se asocia a resistencia insulínica.
Otras adipocinas como la visfatina también se encuentran implicadas en la patogenia del síndrome metabólico. Aun cuando se le considera un origen preferencial por parte del tejido adiposo visceral, la expresión de ARNm de visfatina en tejido visceral y subcutáneo es similar. A pesar de que tiene efecto insulinosensibilizador que actúa sobre el propio receptor de insulina, estimula la producción de IL-1, IL-6 y TNF-α, por lo que se le debe considerar como una citocina proinflamatoria. Está aún por esclarecerse el papel que la visfatina desempeña en la fisiopatología de la obesidad abdominal y el síndrome metabólico.57
En cuanto a la apelina, se ha demostrado el efecto estimulante de la insulina per se sobre su expresión y secreción, tanto in vitro como in vivo. Se ha encontrado una correlación altamente positiva entre la expresión génica de apelina y TNF-α en tejido adiposo de sujetos obesos y delgados.58 La inyección de TNF-α in vivo provoca un aumento tanto de la expresión de apelina en tejido adiposo, como sobre sus niveles circulantes. Se ha observado una correlación positiva entre los niveles de expresión génica de apelina en tejido adiposo subcutáneo y los niveles hepáticos de malondialdehído, un marcador de estrés oxidativo.59 Sin embargo, todavía no está bien establecido si la sobreproducción de apelina ayuda al desarrollo de resistencia insulínica o si, por el contrario, la sobreproducción de apelina podría ser una de las últimas protecciones previas al desarrollo de diabetes tipo 2 y enfermedad cardiovascular.60
La leptina, además de su efecto inhibidor del apetito y estimulador del gasto calórico, tiene efectos promotores de la migración de monocitos y macrófagos, así como de estimuladores de otras citocinas; genera efectos proliferativos, proinflamatorios, protrombóticos y favorecedores del estrés oxidativo, por lo que se la considera uno de los mediadores entre tejido adiposo e inflamación. La estimulación simpática que desencadena la leptina favorece el incremento de la tensión arterial, mientras que el ambiente inflamatorio que generan las quimioquinas y citoquinas de la pared vascular y la síntesis de factores procoagulantes como el inhibidor del plasminógeno tisular (PAI-1), principalmente por la grasa visceral, incrementan el riesgo de enfermedad coronaria. El PAI-1 inhibe la ruptura de coágulo de fibrina favoreciendo la formación de trombos sobre las placas arteroscleróticas rotas.57
Por otra parte, son pocos los estudios que han analizado el papel de la omentina sobre el desarrollo de obesidad y resistencia a la insulina. Se ha demostrado que los niveles de expresión génica en tejido adiposo omental y la secreción de omentina están disminuidos en la obesidad.61 Esta investigación demostró además una correlación negativa entre los niveles circulantes de omentina 1 y el IMC, la circunferencia de cintura, los niveles de leptina y el índice de insulino-resistencia según el modelo de evaluación de la homeostasis (HOMA). Por el contrario, encontró una correlación positiva y significativa entre los niveles plasmáticos de omentina y los niveles circulantes de adiponectina y colesterol HDL.62 Respecto a los mecanismos que regulan la secreción de omentina, un estudio reciente ha descrito que la glucosa y la insulina disminuyen de forma dosis-dependiente la expresión génica, los niveles de proteína y la cantidad de omentina secretada en explantes de tejido adiposo omental.63 Además, la inducción de hiperinsulinemia en sujetos sanos condujo a una reducción de los niveles plasmáticos de omentina -1.62 Todas estas observaciones apuntan a que la disminución en los niveles plasmáticos de omentina podría indicar consecuencias metabólicas o de las comorbilidades asociadas con la obesidad.
La resistina no es una verdadera adipocina pues, aunque se produce en el tejido adiposo, su origen se relaciona con la fracción estromal y no con los adipocitos. Aunque en animales de experimentación se ha visto implicada en la fisiopatología de la IR, no hay tal evidencia en humanos, y se observa relacionada con la inflamación y sus marcadores biológicos.
Otras citocinas también participan en la fisiopatología de la inflamación. La IL-10, secretada por células inmunitarias, parece tener efecto protector contra la disfunción endotelial y la arteriosclerosis. La disminución de IL-10 facilita la inestabilidad de la placa arteriosclerótica y el desarrollo de síndrome metabólico.
Diversas alteraciones metabólicas relacionadas con la obesidad abdominal pueden explicarse por una producción excesiva y el consiguiente aporte al hígado, a través de la vena porta, de AGL procedentes del TAV. En principio, la magnitud de la grasa visceral y el flujo de AGL que alcanza el hígado son proporcionales. La concentración plasmática de AGL es un 20 % mayor en obesos que en no obesos, y la contribución de la lipólisis esplácnica a la llegada de AGL al hígado es función del volumen de grasa visceral, aspecto más significativo en mujeres. Esta es una de las razones por las que el tejido adiposo visceral es un mejor predictor (con respecto al tejido subcutáneo) de las complicaciones metabólicas de las comorbilidades de la obesidad.
Los adipocitos hipertróficos del compartimento visceral son resistentes al efecto antilipolítico de la insulina, lo que favorece el aumento de la salida de AGL vía portal y origina en el hepatocito un aumento de secreción de lipoproteínas ricas en triglicéridos y reducción de la degradación de apoproteína B y de la extracción hepática de insulina. La transferencia de triglicéridos a partículas de lipoproteínas de baja densidad (LDL) y lipoproteínas de alta densidad (HDL) y su ulterior lipólisis por la lipasa hepática da lugar a partículas HDL y LDL pequeñas, y a disminución de la concentración de HDL, configurando la dislipemia aterogénica que acompaña al síndrome metabólico. El flujo aumentado de AGL participa en la fisiopatología de la esteatosis no sólo hepática y muscular, sino también cardiaca y pancreática, lo que produce las consiguientes alteraciones funcionales.63
Además de promover la inflamación de bajo grado y la IR, la excesiva producción de AGL y de adipocinas por el TAV participa en otros mecanismos fisiopatológicos que contribuyen a incrementar el riesgo cardiovascular.64
La obesidad abdominal se asocia a hepatopatía grasa no etílica (HGNE), que se produce por infiltración adiposa de los hepatocitos, en la que el flujo incrementado de AGL procedente de la grasa visceral tiene un papel significativo. Independientemente de su asociación con el síndrome metabólico, la HGNE puede contribuir por sí sola a favorecer el riesgo cardiovascular y metabólico generando IR, dislipemia, inflamación y estrés oxidativo, que a su vez se ven implicados en el deterioro de la función hepática, con lo que se cierra un círculo vicioso.65 La reducción de adiponectina se relaciona con la gravedad histológica independientemente de la obesidad abdominal, o de los componentes del síndrome metabólico. Análogamente al depósito de grasa en el hígado, el páncreas y el músculo esquelético, la frecuente disfunción cardiaca que acompaña a la obesidad puede estar, al menos parcialmente, relacionada con el depósito intramiocárdico de triglicéridos, que es 5-6 veces superior que en sujetos no obesos, y favorecer así la aparición de insuficiencia cardiaca. (Figura 1). En consonancia con estos datos, el tratamiento de la obesidad mediante derivación gástrica reduce la masa ventricular y mejora su función.66
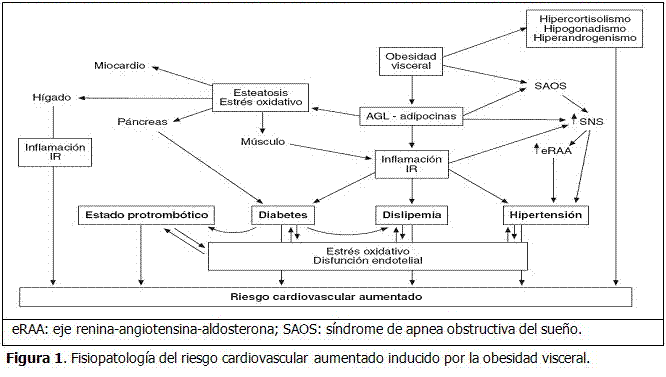
El depósito adiposo centrípeto se asocia a síndrome de apneas obstructivas del sueño, demás se ha mostrado ligada a otros procesos patológicos como diferentes tipos cáncer y la infertilidad.
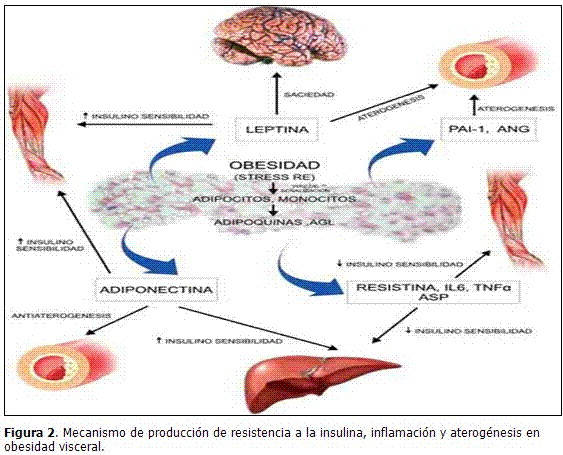
La obesidad produce estrés del retículo endoplásmico, que conlleva a la activación de vías de señalización que aumentan la producción de adipoquinas, la insulino resistencia y la liberación de ácidos grasos libres. Estos últimos estimulan los receptores tipo Toll de los monocitos que sintetizan TNF-α e IL-6, y que se unen a los adipocitos, manteniendo la lipólisis y promoviendo la participación de otras vías de señalización que modulan la insulino sensibilidad, la síntesis de adipoquinas y un aumento en la migración de más monocitos, para perpetuar el estado inflamatorio. (Figura 2).27
CONCLUSIONES
El tejido adiposo es un órgano con importante función endocrina, capaz de secretar diversas sustancias conocidas en general como adipocinas, entre ellas, la adiponectina, angiotensinógeno, leptina, resistina, factor de necrosis tumoral alfa, interleucina 6, visfatina, omentina, apelina, resistina, proteína 4 ligante de retinol, ácidos grasos no esterificados, inhibidor del activador del plasminógeno, adipsina y proteína estimulante de acetilación. Todas ellas, con profundas implicaciones en la homeostasis de la glucosa, inciden también en la regulación del peso corporal, en el sistema inmune, en la función vascular y en la sensibilidad insulínica o insulinorresistencia. En la obesidad visceral se produce un estado proinflamatorio que tiene una relación bidireccional con la insulinoresistencia, que contribuye a generar hipertensión, dislipemia, disglucosis y una situación protrombótica que refuerza la morbimortalidad cardiovascular.
REFERENCIAS BIBLIOGRÁFICAS
- Vögler O, López-Bellan A, Alemany R, Tofé S, González M, Quevedo J, et al. Structure-effect relation of C18 long-chain fatty acids in the reduction of body weight in rats. International Journal of Obesity. 2008;32(3):451-63 [Buscar en Google Scholar]
- Wong GW, Wang J, Hug C, Tsao S, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci USA. 2004;101(28):10302-07 [Buscar en Google Scholar]
- Simha V, Szczepaniak LS, Wagner AJ, DePaoli AM, Garg A. Effecto of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes Care. 2003;26(1):30-35 [Buscar en Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin invest. 2000;106(2):171-6 [Buscar en Google Scholar]
- Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Cliln Endocrinol. 2006;64(4):355-65 [Buscar en Google Scholar]
- Yki-Jarvinen, H. Ectopic fat accumulation: an important cause of insulin resistance in humans. J R Soc Med. 2002;95:39-45 [Buscar en Google Scholar]
- Argente J, Martos-Moreno Ga, Hernández M. Mesa Redonda: El tejido adiposo como glándula endocrina. Obesidad y síndrome metabólico. Bol Pediatr. 2006;46:269-74 [Buscar en Google Scholar]
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucosa homeostasis. Nature. 2006;444(5483):847-53 [Buscar en Google Scholar]
- Hausman GJ. Identification of adipose tissue primordia in perirenal tissues of pig fetuses: utility of phosphatase histochemistry. Acta Anat (Basel). 1987;128(3):236-42 [Buscar en Google Scholar]
- Vázquez Vela ME, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Arch Med Res. 2008;39(8):715-28 [Buscar en Google Scholar]
- Cinti S. The role of brown adipose tissue in human obesity. Nutr Metab Cardiovasc Dis. 2006;16(8):569-74 [Buscar en Google Scholar]
- Valenzuela A, Sanhueza J. El tejido adiposo: algo más que un reservorio de energía. Grasas y Aceites. 2009;60(5):439-52 [Buscar en Google Scholar]
- Garaulet M, Hernandez Morante JJ, Lujan J, Tebar FJ, Zamora S. Relationship between fat cell size and number and fatty acid composition in adipose tissue from different fat depots in overweight/obese humans. Int. J. Obes. 2006;30(6):899-905 [Buscar en Google Scholar]
- Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, et al. 2004. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem. 2004;279(40):42062-71 [Buscar en Google Scholar]
- Sethi JK, Vidal Puig AJ. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res. 2007;48(6):1253-62 [Buscar en Google Scholar]
- Tchernof A, Belanger C, Morisset AS, Richard C, Mailloux J, Laberge P, et al. Regional differences in adipose tissue metabolism in women. Minor effect of obesity and body fat distribution. Diabetes. 2006;55(5):1353-60 [Buscar en Google Scholar]
- Kuk JL, Ross R. Influence of sex on total and regional fat loss in overweight and obese men and women. Int J Obes. 2009;33(6):629-34 [Buscar en Google Scholar]
- Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, et al. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation. 2007;116(11):1234-41 [Buscar en Google Scholar]
- Dolinkova M, Dostalova I, Lacinova Z, Michalsky D, Haluzikova D, Mraz M, et al. The endocrine profile of subcutaneous and visceral adipose tissue of obese patients. Mol Cell Endocrinol. 2008;291(1-2):63-70 [Buscar en Google Scholar]
- Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, et al. CC chemokine and CC chemokine receptorprofiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab. 2008;93(8):3215-21 [Buscar en Google Scholar]
- Bluher M, Williams CJ, Kloting N, His A, Ruschke K, Oberbasch A, et al. Gene expression of adiponectin receptors in human visceral and subcutaneous adipose tissue is related to insulin resistance and metabolic parameters and is altered in response to physical training. Diabetes Care. 2007;30(12):3110-5 [Buscar en Google Scholar]
- Hou M, Xia M, Zhu H, Wang Q, Li Y, Xiao Y et al. Lysophosphatidylcholine promotes cholesterol efflux from mouse macrophage foam cells via PPARγ-LXRα-ABCA1-dependent pathway associated with apoE. Cell Biochem Funct. 2007;25(1):33-44 [Buscar en Google Scholar]
- Escribá PV. Membrane-lipid therapy: a new approach in molecular medicine. Trends Mol Med. 2006;12(1):34-43 [Buscar en Google Scholar]
- Inokuma, K, Okamatsu-Ogura Y, Omachi A, Matsushita Y, Kimura K, Yamashita H, et al. Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. AJP Endo. 2006;290(5):E1014-21 [Buscar en Google Scholar]
- Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105-20 [Buscar en Google Scholar]
- Tchernof A, Desmeules A, Richard C, Laberge P, Daris M, Mailloux J, et al. Ovarian hormone status and abdominal visceral adipose tissue metabolism. J Clin Endocrinol Metab. 2004;89(7):3425-30 [Buscar en Google Scholar]
- Pérez Mayorga M. El adipocito como órgano endocrino. Implicaciones fisiopatológicas y terapéuticas. Rev. Med. 2007;15(2):225-42 [Buscar en Google Scholar]
- Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr. Rev. 2002;60 Suppl 10:1-14 [Buscar en Google Scholar]
- Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145 Suppl 5:2273-82 [Buscar en Google Scholar]
- Bjorbaek C, Kahn BB. Leptin signaling in the central nervous system and the periphery. Prog Horm Res. 2004;59(1):305-31 [Buscar en Google Scholar]
- Morales Villegas E. Síndrome X vs síndrome metabólico: Entendiendo sus coincidencias y sus directas hacia una “nueva cardiología”. Arch Cardiol Méx. 2006;76 Suppl 4:173-88 [Buscar en Google Scholar]
- Sánchez Muñoz F, García Macedo R, Alarcón Aguilar F, Cruz M. Adipocinas, tejido adiposo y su relación con células del sistema inmune. Gac Méd Méx. 2005;141(6):505-12 [Buscar en Google Scholar]
- Szmitko PE, Teoh H, Stewart D, Verma S. Adiponectin and cardiovascular disease: state of the art?. Am J Physiol Heart Circ Physiol. 2007;292(4):1655-63 [Buscar en Google Scholar]
- Cersosimo E, DeFronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. 2006;22(6):423-36 [Buscar en Google Scholar]
- Strawbridge AB, Elmendorf JS. Endothelin-1 impairs glucose transporter trafficking via a membrane based mechanism. J Cell Biochem. 2006;97(4):849-56 [Buscar en Google Scholar]
- Kim JA, Montagnani M, Kon KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction. Molecular and pathophysiological mechanisms. Circulation. 2006;113(15):1888-904 [Buscar en Google Scholar]
- Salvador J. The debate about metabolic syndrome: a cluster of concepts, perspectives, semantics and research. Obes Metab. 2007;3:1-4 [Buscar en Google Scholar]
- Kershaw E, Flier J. Adipose Tissue as an Endocrine Organ. J Clin Endocrinol Met. 2004;89(6):2548-56 [Buscar en Google Scholar]
- Bletowski J. Apelin and visfatin: Unique “beneficial” adipokines upregulated in obesity?. Med Sci Monit. 2006;12(6):112-19 [Buscar en Google Scholar]
- Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M, et al. Hormonal regulation of the novel adipocytokine visfatinin 3T3-L1 adipocytes. J Endocrinol. 2005;185(3):R1-8 [Buscar en Google Scholar]
- Kralisch S, Klein J, Lossner U. Bluher M, Paschke R, Stumvoll M, Fasshauer M. Interleukine-6 is a negative regulator of visfatin gene expression in 3t3L-1 adipocites. Am J Physiol Endrocrinol Metab. 2005;289(4):E586-90 [Buscar en Google Scholar]
- Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depotspecific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290(6):E1253-E61 [Buscar en Google Scholar]
- Lee DK, Valdivia VR, Nguyen T, Cheng R, George SR, O Dowd BF et al. Modification of the terminal residue of apelin 13 antagonizes its hypotensive action. Endocrinology. 2005;146(1):231-36 [Buscar en Google Scholar]
- Lee CR, Watkins ML, Patterson JH, Gattis W, O connor CM, Gheorghiade M et al. Vasopressin: a new target for the treatment of heart failure. Am Heart J. 2003;146(1):9-18 [Buscar en Google Scholar]
- Jaszberenyi M, Bujdoso E, Telegdy G. Behavioral, neuroendocrine and thermoregulatory actions of apelin-13. Neuroscience. 2004;129(3):811-16 [Buscar en Google Scholar]
- Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Arder DI. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111(7):932-39 [Buscar en Google Scholar]
- Graham T, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354(25):2552-63 [Buscar en Google Scholar]
- Brekke M, Gjelsvik B. Secondary cardiovascular risk prevention -we can do better. Lancet. 2009;373(9667):873-5 [Buscar en Google Scholar]
- Gudbrandsen OA, Gudbrandsen OA, Hultstrøm M, Leh S, Monica Bivol L, Vagnes O, et al. Prevention of hypertension and organ damage in 2-kidney, 1-clip rats by tetradecylthioaceticacid. Hypertension. 2006;48(3):460-6 [Buscar en Google Scholar]
- Goldfine A, Beckman J, Betensky R, Devlin H, Hurley S, Varo N, et al. Family history of diabetes is a major determinant of endothelial function. J Am Coll Cardiol. 2006;47(12):2456-61 [Buscar en Google Scholar]
- Lobby P. Changing concepts of atherogenesis. J Intern Med. 2000;247(3):349-58 [Buscar en Google Scholar]
- Skurk T, Alberti Huber C, Herder C, Hauner H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J Clin Endocrinol Metab. 2007;92(3):1023-33 [Buscar en Google Scholar]
- Charriere G, Cousin B, Arnaud E, Andre M, Bacou F, Penicaud L, et al. Preadipocyte conversion to macrophage. Evidence of plasticity. J Biol Chem. 2003;278(11):9850-55 [Buscar en Google Scholar]
- Matsushita K, Yatsuya H, Tamakoshi K, Wada K, Otsuka R, Takefuji S, et al. Comparison of circulating adiponectin and proinflammatory markers regarding their association with metabolic syndrome in Japanese men. Arterioscler Thromb Vasc Biol. 2006;26(4):871-6 [Buscar en Google Scholar]
- Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106(22):2767-70 [Buscar en Google Scholar]
- Domínguez Reyes CA. Adiponectina. El tejido adiposo más allá de la reserva inerte de energía. Revista de Endocrinología y Nutrición. 2007;15(3):149-55 [Buscar en Google Scholar]
- Harle P, Straub RH. Leptin is a link between adipose tissue and inflammation. Ann N Y Acad Sci. 2006;1069:454-62 [Buscar en Google Scholar]
- Daviaud D, Boucher J, Gesta S, Dray C, Guigne C, Quilliot D, et al. TNF alpha up-regulates apelin expression in human and mouse adipose tissue. FASEB J. 2006;20(9):1528-30 [Buscar en Google Scholar]
- García Díaz D, Campión J, Milagro FI, Martínez JA. Adiposity dependent apelin gene expression: relationships with oxidative and inflammation markers. Mol Cell Biochem. 2007;305(1-2):87-94 [Buscar en Google Scholar]
- Carpéné C, Dray C, Attané C, Valet P, Portillo MP, Churruca I, et al. Expanding role for the apelin/APJ system in physiopathology. J Physiol Biochem. 2007;63:359-73 [Buscar en Google Scholar]
- De Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes. 2007;56(6):1655-61 [Buscar en Google Scholar]
- Tan BK, Adya R, Farhatullah S, Lewandowski KC, O Hare P, Lehnert H, et al. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. 2008;57(4):801-8 [Buscar en Google Scholar]
- Després JP. Is visceral obesity the cause of the metabolic syndrome?. Ann Med. 2006;38(1):52-63 [Buscar en Google Scholar]
- McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40 Suppl 1:17-29 [Buscar en Google Scholar]
- Targher G, Bertolini L, Rodella S, Zoppini G, Scala L, Zenari L, et al. Associations between plasma adiponectin concentrations and liver histology in patients with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf). 2006;64(6):679-83 [Buscar en Google Scholar]
- Giovannucci E. Metabolic syndrome, hyperinsulinemia, and colon cancer: a review. Am J Clin Nutr. 2007;86(3):S836-42 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129