
Presentaciones de casos
Síndrome de Morris completo. Presentación de un caso
Complete Morris Syndrome. Case Presentation
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2020-01-30 14:53:28
Aprobado: 2020-02-06 10:03:49
Correspondencia: Manyeles Brito Vázquez. Hospital Pediátrico Docente Provincial José Martí Pérez. Sancti Spíritus. manyeles.ssp@infomed.sld.cu
RESUMEN
Palabras clave: diferenciación sexual; hernia inguinal; presentación de caso
ABSTRACT
Key words: sexual differentiation; inguinal hernia; case presentation
INTRODUCCIÓN
El síndrome de insensibilidad androgénica (SIA), síndrome de Morris o feminización testicular, es un desorden en la diferenciación sexual XY, en el que los órganos diana son incapaces de reaccionar a la testosterona (T) o dihidrotestosterona (DHT). Como consecuencia, aunque las gónadas son testículos, los genitales no se virilizan adecuadamente durante el desarrollo fetal y los individuos tienen una apariencia externa femenina. La falla en la acción de los andrógenos se debe a una alteración del receptor, siendo este el mecanismo fisiopatológico esencial del síndrome de insensibilidad a andrógenos (SIA).(1,2)
La etiopatogenia reside en mutaciones del gen localizado en el locus Xq11-12 del brazo largo del cromosoma X que codifica los receptores intracelulares de andrógenos y que provoca una ausencia de estímulo y respuesta de los órganos diana a dichas hormonas. Aproximadamente 2/3 de los casos se transmiten de modo recesivo ligado al cromosoma X y 1/3 corresponde a mutaciones de novo.(1,3-7) Hasta la fecha, la base de datos de las mutaciones del gen receptor de andrógenos, ha reportado más de 800 diferentes mutaciones en pacientes con SIA.(4,6)
Si la falla del receptor es severa se produce la forma completa de SIA, las formas menos severas se caracterizan por virilización incompleta, con o sin infertilidad y las formas con moderada severidad se caracterizan por grados diversos de ambigüedad sexual.(1,3)
La forma completa de SIA es relativamente rara. En la infancia la presentación clínica más común es la aparición de hernia inguinal bilateral. Los individuos no diagnosticados durante la infancia son detectados después de la pubertad debido a amenorrea primaria.(1-5,7,8)
Epidemiológicamente, el SIA es una enfermedad rara y se estima un caso de SIA completo por cada 20,000 a 64,000 recién nacidos varones(1,8,9) aunque existen otras estadísticas que plantean una prevalencia de 1:20 400 a 1:99 100 recién nacidos genéticamente masculinos.(3-5)
Se presenta el caso por lo poco frecuente de la aparición de esta entidad.
PRESENTACIÓN DEL CASO
Se presenta el caso de una escolar de 9 años de edad, nacida de un embarazo normal, parto distócico por cesárea a las 37 semanas de estado gestacional, sin infecciones interrecurrentes, ni radiaciones, la fenilcetonuria al 5to día negativa.
Se le realizó interconsulta a los 2 meses de edad con el Servicio de Cirugía Pediátrica porque al cambiar el pañal la madre notó que protruía un aumento de volumen en ambos canales inguinales, interpretándose como hernia inguinal bilateral. Se decidiórealizar intervención quirúrgica y en el acto se constataron gónadas con aspecto macroscópico de testes; se realizó toma de muestra para biopsia y se corroboró la presencia de testículos mediante anatomía patológica (08-B-83).
Ante la sospecha de un desorden en la diferenciación sexual se realizó cariotipo, describiéndose como cromosómicamente normal con sexo cromosómico masculino (cariotipo 46XY, 12 metafases).
La paciente regresó a los 9 años buscando reevaluación por las especialidades de endocrinología y cirugía pediátrica.
Examen físico
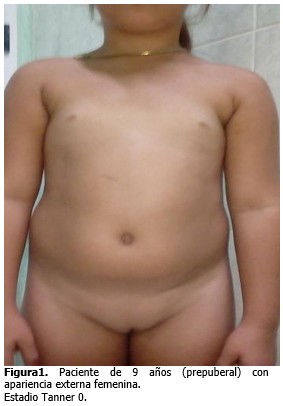
-Mamas: escala de Tanner 0.
-Vello pubiano: escala Tanner 0.
-Palpación de testes en ambas regiones inguinales.
-Genitales externos de apariencia femenina, con clítoris y labios menores hipotróficos. (Figura 1).
Perfil hormonal
TSH: 4mui/L.
17OHP: 4,44 ng/ml.
Testosterona: 8 ng/ml / elevada.
FSH: 25,5 nmol/L/ elevada.
LH: 2,9 nmol/L.
Prolactina: 549 nmol/L.
Ultrasonido ginecológico
No se observó imagen sugestiva de cuerpo uterino, aunque se constató la vagina hasta su tercio medio. No se constataron anejos.
Se exploró con transductor de partes blandas ambos canales inguinales, llamó la atención a nivel del canal derecho imagen de baja ecogenicidad, fusiforme, de 20x15mm, que impresionaba en el interior del anillo inguinal por la cercanía a los vasos ilíacos y que en una exploración dinámica protruía hacia la pared anterior con maniobra de Valsalva. En la región inguinal izquierda se mostró una imagen similar de 29x9.2 mm que se mantenía fija sin movilizarse con dicha maniobra.
Resonancia magnética nuclear (RMN) de pelvis
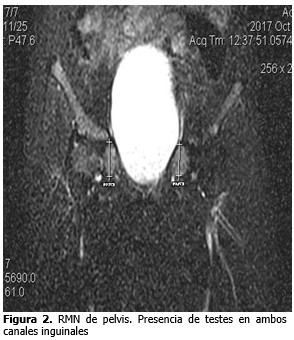
Se observaron en canales inguinales en ambos testes, el derecho medía 2,2 cm.el izquierdo 2,8 cm. homogéneo. (Figura 2).
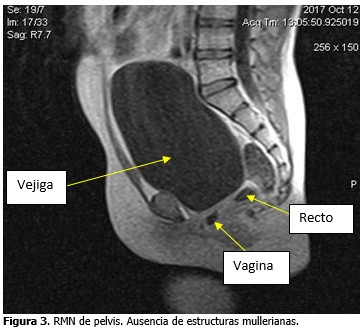
Se observó ausencia de útero y anejos. Permeabilidad a nivel de los genitales externos, que abocan en un saco ciego en el interior de la pelvis. (Figura 3).
Luego de su evaluación en consulta multidisciplinaria, se realizó orquiectomía por vía laparoscópica, extrayéndose los testes, sin la realización por el momento de plastia vaginal. La paciente tuvo un posoperatorio adecuado y ahora se encuentra con terapia hormonal de reemplazo.
DISCUSIÓN
El síndrome de insensibilidad a los andrógenos fue descrito por primera vez por Morris en 1953, con la descripción clínica de 82 pacientes con testes pero fenotipo femenino, razón por la cual Morris nombró este síndrome como “feminización testicular”.(5,7,10)
El diagnóstico se sospecha generalmente cuando el individuo acude por hernia inguinal bilateral en la infancia, como ocurrió en este caso o por una amenorrea primaria después de la pubertad.(1-5,7,8) No obstante, existen reportes de casos que se presentan por infertilidad después del matrimonio e incluso con tumores del aparato reproductor como carcinoma in situ y seminoma testicular.(1, 3-7)
La forma total del SIA se caracteriza por:
- Ausencia de epidídimos, vasos deferentes y vesículas seminales, estructuras derivadas de los conductos de Wolff, que no responden al estímulo de la testosterona producida por las células de Leydig.
- Ausencia de próstata y genitales externos (pene, uretra peneana y bolsas escrotales) al necesitar el seno y el tubérculo urogenital de la estimulación por parte de la dihidrotestosterona (DHT) para su desarrollo.
- Ausencia de trompas de Falopio, útero, cérvix y tercio superior de la vagina debido a que las células de Sertoli segregan con normalidad la hormona antimülleriana (HAM) que evitará el desarrollo de estas estructuras, dependientes de los conductos müllerianos.
- Presencia de testículos no descendidos (frecuentemente criptorquídicos localizados en anillo inguinal, intra-abdominales o a nivel labial) ya que la HAM provoca el descenso transabdominal de los testes pero no alcanzan el pliegue labio-escrotal debido a la ausencia de la estimulación androgénica.
- Vagina corta con extremo ciego al desarrollarse solo la porción inferior derivada del seno urogenital, clítoris y labios mayores normales, labios menores hipotróficos y ausencia de himen.
- Desarrollo normal de las glándulas mamarias debido a los estrógenos segregados por los testículos y por la conversión periférica de los andrógenos, pero con hipopigmentación de las areolas y pezones de pequeño tamaño.
- Disminución de la cantidad de vello pubiano y axilar.(2,5,7,8,10)
Existe una forma parcial de insensibilidad en la que se pueden ver manifestaciones androgénicas de intensidad variable.(1-10)
El perfil hormonal suele mostrar LH elevada, lo que refleja la falta de respuesta hipotalámica a la testosterona, la cual muestra niveles normales o ligeramente elevados. Los niveles de estradiol serán elevados como un efecto directo de la secreción testicular y la aromatización periférica de los andrógenos con la consiguiente conversión en estrógenos y la FSH se encontrará de normal a elevada.(3-5,7) En el caso presentado la dosificación sérica de testosterona y FSH fue elevada, con el resto del perfil hormonal realizado dentro de parámetros normales. El nivel elevado de HAM puede ser un marcador de resistencia a los andrógenos en el primer año de vida y después del comienzo de la pubertad.(5)
Las imágenes radiológicas juegan un importante papel en el diagnóstico y manejo del SIA. La ultrasonografía es la primera línea de investigación para confirmar la agenesia mulleriana y la localización de los testes, específicamente al nivel del canal inguinal o los labios mayores. No obstante, ya que es un operador dependiente y puede ser inconclusa, la resonancia magnética nuclear (RMN) es el examen por excelencia, con un 100% de precisión, para el diagnóstico de la agenesia mulleriana, el hallazgo de los remanentes wolffianos, la evaluación de la terminación ciega de la vagina, la localización pélvica o abdominal de los testes, así como el desarrollo de cualquier proceso maligno gonadal. Las imágenes sagitales y transversas potenciadas en T1 y T2 son las más usadas con estos fines.(2,7,9,10)
El abordaje de esta entidad debe realizarse en forma multidisciplinaria e incluir la gonadectomía y posterior reemplazo hormonal, la creación de una vagina funcional, el asesoramiento genético y el apoyo psicológico.(1-5)
El manejo estándar del SIA es la gonadectomía, de preferencia por vía laparoscópica, para prevenir posibles transformaciones malignas en los testes. Después de la gonadectomía la terapia estrogénica de reemplazo es indispensable para mantenerla feminidad y proveer una adecuada mineralización ósea. (1-3,5,7) Las opciones para la creación vaginal incluyen la dilatación o creación quirúrgica.(1, 3, 5)
El síndrome de insensibilidad androgénica (SIA) es un desorden en la diferenciación sexual XY producido por una falla en la acción de los andrógenos debido a una alteración de sus receptores. Esta entidad debe ser considerada en pacientes que presenten hernia inguinal en la infancia, como en el presente caso, amenorrea primaria o variaciones en la apariencia de los genitales externos, en el caso de las formas incompletas. Las pruebas de imagen juegan un importante rol en la identificación de los testes ectópicos. El tratamiento debe ser integral y debe incluir la gonadectomía, terapia hormonal de reemplazo, cirugía reconstructiva vaginal y la intervención psicológica.
Conflicto de intereses: los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Contribución de los autores:
Idea conceptual: Manyeles Brito Vázquez.
Análisis estadístico: Ángela Belkis Brito García.
Revisión de la literatura: Manyeles Brito Vázquez, Ángela Brito García, Delvis Batista García
Escritura del artículo: Manyeles Brito Vázquez.
Revisión crítica del artículo: Delvis Batista García.
Financiación: Hospital Pediátrico Docente Provincial José Martí Pérez. Sancti Spíritus.
REFERENCIAS BIBLIOGRÁFICAS
- Gómez V, Amies AM. Disorders of Sexual Development in Adult Women. Obstet Gynecol. 2016;128(5):1162-73 [Buscar en Google Scholar]
- Souhail R, Amine S, Nadia A, Tarik K, Khalid EK, Abdellatif K, et al. Complete androgen insensitivity syndrome or testicular feminization: review of literature based on a case report. Pan Afr Med J. 2016;25(1):199 [Buscar en Google Scholar]
- Mongan NP, Tadokoro R, Bunch T, Hughes LA. Androgen insensitivity syndrome. Best Practice and Research. Clinic Endocrinol Metab. 2015;29(4):569-80 [Buscar en Google Scholar]
- CemiK, Serdar S, Gonul C, Husseyin O, Bumin N. A Novel Mutation in Human Androgen Receptor Gene Causing Partial Androgen Insensitivity Syndrome in a Patient Presenting with Gynecomastia at Puberty. J Clin Res Pediatr Endocrinol. 2016;8(2):232-5 [Buscar en Google Scholar]
- Batista Rl, Costa EMF, Rodríguez AS, Gómez NL, Faria Jr, Nishi MY, et al. Androgen insensitivity syndrome: a review. Arch Endocrinol Metab. 2018;62(2):227-35 [Buscar en Google Scholar]
- Touzon MS, Pérez N, Marino R, Ramirez P, Constanzo M, Guercio G, et al. Androgen Insensitivity Syndrome: Clinical Phenotype and Molecular Analysis in a Single Tertiary Center Cohort. J Clin Res Pediatr Endocrinol. 2019;11(1):24-33 [Buscar en Google Scholar]
- Baca JE, Leuchter IJ, Domínguez CLG, Mayagoitia GJC. Testicular torsion in the inguinal canal with recurrent hernia, in a patient with complete androgenic insensitivity syndrome («feminizing testicle»). Rev Mex Cir Endosc. 2018;19(2):82-6 [Buscar en Google Scholar]
- Borrego JA, Varona JA, Areces G, Formoso LE. Síndrome de Morris. Rev Cubana Obst Ginecol [revista en Internet]. 2012 [citado 17 Mar 2018];38(3):[aprox. 10p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_abstract&pid=S0138-600X2012000300014&lng=es&nrm=iso [Buscar en Google Scholar]
- Skiker I. Complete androgen sensitivity syndrome. Diagnostic and Interventional Imaging. 2015;96(1):121-5 [Buscar en Google Scholar]
- Samy AM. Androgen insensitivity syndrome with inguinal testes: MRI diagnosis. Egyp J Radiol Nuc Med. 2016;47(1):607-9 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129